Pelc LA, Chen Z, Gohara DW, Vogt AD, Pozzi N, Di Cera E
Biochemistry 2015 Feb;54(7):1457-64
PMID: 25664608
Abstract
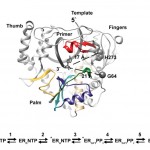
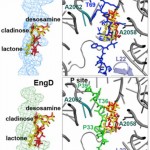
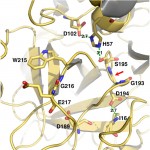
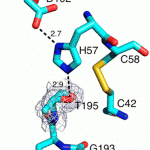
Although Thr is equally represented as Ser in the human genome and as a nucleophile is as good as Ser, it is never found in the active site of the large family of trypsin-like proteases that utilize the Asp/His/Ser triad. The molecular basis of the preference of Ser over Thr in the trypsin fold was investigated with X-ray structures of the thrombin mutant S195T free and bound to an irreversible active site inhibitor. In the free form, the methyl group of T195 is oriented toward the incoming substrate in a conformation seemingly incompatible with productive binding. In the bound form, the side chain of T195 is reoriented for efficient substrate acylation without causing steric clash within the active site. Rapid kinetics prove that this change is due to selection of an active conformation from a preexisting ensemble of reactive and unreactive rotamers whose relative distribution determines the level of activity of the protease. Consistent with these observations, the S195T substitution is associated with a weak yet finite activity that allows identification of an unanticipated important role for S195 as the end point of allosteric transduction in the trypsin fold. The S195T mutation abrogates the Na(+)-dependent enhancement of catalytic activity in thrombin, activated protein C, and factor Xa and significantly weakens the physiologically important allosteric effects of thrombomodulin on thrombin and of cofactor Va on factor Xa. The evolutionary selection of Ser over Thr in trypsin-like proteases was therefore driven by the need for high catalytic activity and efficient allosteric regulation.